Mit dem Ende der Übergangsfrist und dem Geltungsbeginn der Medical Device Regulation (745/2017 (EU)), am 26.05.2021 werden die Pflichten und Aufgaben aller Akteure, die Medizinprodukte herstellen und vertreiben, neu reguliert. Ein Jahr später, am 26. Mai 2022, zieht dann auch die Verordnung für in-Vitro-Diagnostika 746/2017 (EU) nach. Bestehende Medizinprodukte, deren Konformität noch nach MDD/IVDD bescheinigt wurde, dürfen bis maximal 27.05.2025 ohne neue Zertifizierung weiter bereitgestellt werden. Aber auch über die Neuzulassung von Produkten hinaus kommen auf Hersteller und Inverkehrbringer von Medizinprodukten ab dem 26.05.2021 neue Aufgaben zu.
Neben mehr Verbindlichkeit bei den Anforderungen auf internationaler Ebene, liegt der Fokus der Umstellung auf der EU-weiten Vereinheitlichung der Pflichten und auf mehr Transparenz im europäischen Wirtschaftsraum. So müssen künftig alle Beteiligten eindeutig definieren, ob sie als Hersteller, Bevollmächtigter, Händler oder Importeur auf dem Markt auftreten. Dies erfolgt über die Registrierung bei der zentralen Datenbank EUDAMED und die Zuweisung einer Single Registration Number (SRN). Auch für alle Medizinprodukte wird in der MDR die eindeutige Identifikation und Rückverfolgbarkeit anhand einer Registrierung bei der EUDAMED gefordert. Die Kennzeichnung erfolgt durch Aufbringen einer Unique Device Identifiation (UDI) number auf jedes einzelne in Verkehr gebrachte Produkt.
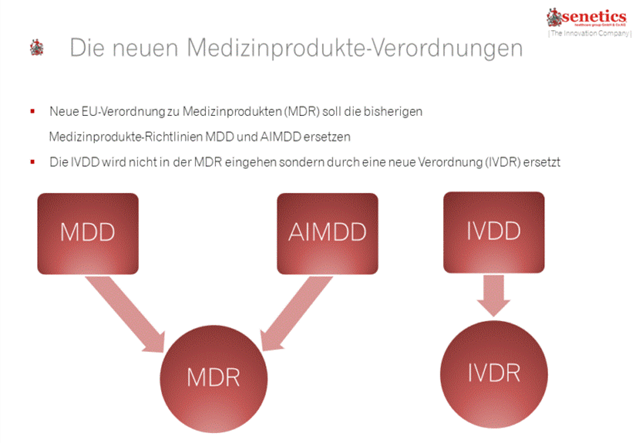
Grundsätzlich ist der Tonus der MDR jedoch ähnlich zu den früheren Medizinprodukte-Richtlinien MDD, AIMDD und IVDD:
Medizinprodukte und in-vitro Diagnostika durchlaufen ein Konformitätsbewertungsverfahren, deren Ziel eine Konformitätserklärung darstellt, die in der CE Kennzeichnung mündet. Hierbei bleiben die Rahmenbedingungen ähnlich: Basierend auf der Zweckbestimmung werden Medizinprodukte und in-vitro Diagnostika klassifiziert, wodurch die Anforderungen an das jeweilige Produkt abgeleitet werden können.